-
实时荧光定量PCR 编辑
所谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
DNA结合染料法 | 基于探针的化学法 | 猝灭染料引物法 | |
|---|---|---|---|
举例 | SYBR Green I | TaqMan、分子信标、Scorpion和杂交探针 | Amplifluor和LUX荧光引物 |
基本原理 | 应用一种带有荧光的、非特异的DNA结合染料检测PCR过程中积累的扩增产物。 | 采用荧光标记引物扩增,从而使荧光标记基团直接掺入PCR扩增产物中;依赖荧光能量共振传递(FRET)。 | |
特异性 | 仅检测特异性扩增产物。 | 检测特异性扩增产物及非特异反应产物,如引物二聚体。 | |
应用 | DNA及RNA定量;基因表达验证 | ||
优点 | 可对任何双链DNA进行定量;不需要探针,因此减少了实验设计及运转成本;适合于大量基因的分析;简单易用。 | ||
缺点 | 对于不同的靶序列需要合成不同的探针,原料成本较高。 | ||
检测方法
1.SYBRGreenⅠ法:
在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。
SYBR定量PCR扩增荧光曲线图
PCR产物熔解曲线图(单一峰图表明PCR扩增产物的单一性)
2.TaqMan探针法:
探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5’-3’外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物的形成完全同步。
技术原理
将标记有荧光素的Taqman探针与模板DNA混合后,完成高温变性,低温复性,适温延伸的热循环,并遵守聚合酶链反应规律,与模板DNA互补配对的Taqman探针被切断,荧光素游离于反应体系中,在特定光激发下发出荧光,随着循环次数的增加,被扩增的目的基因片段呈指数规律增长,通过实时检测与之对应的随扩增而变化荧光信号强度,求得CT值,同时利用数个已知模板浓度的标准品作对照,即可得出待测标本目的基因的拷贝数。
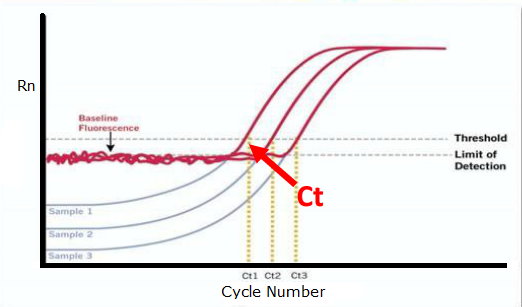
Ct 值
Ct值(Cycle threshold,循环阈值)的含义为:每个反应管内的荧光信号到达设定阈值时所经历的循环数
 图1
图1
(如图1所示)。
1. 荧光阈值(threshold)的设定
PCR反应的前15个循环的荧光信号作为荧光本底信号,荧光阈值的缺省(默认)设置是3-15个循环的荧光信号的标准偏差的10倍,即:threshold = 10*SDcycle 3-15
2. Ct值与起始模板的关系
每个模板的Ct值与该模板的起始拷贝数的对数存在线性关系,公式如下。
Ct=-1/lg(1+Ex)*lgX0+lgN/lg(1+Ex)
n为扩增反应的循环次数,X0为初始模板量,Ex为扩增效率,N为荧光扩增信号达到阈值强度时扩增产物的量。
起始拷贝数越多,Ct值越小。利用已知起始拷贝数的标准品可作出标准曲线,其中横坐标代表起始拷贝数的对数,纵坐标代Ct值。因此,只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。
荧光化学物质
实时荧光定量PCR所使用的荧光物质可分为两种:荧光探针和荧光染料。现将其原理简述如下:
1. TaqMan荧光探针:PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5'-3'外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。而新型TaqMan-MGB探针使该技术既可进行基因定量分析,又可分析基因突变(SNP),有望成为基因诊断和个体化用药分析的首选技术平台。
2. SYBR荧光染料:在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料非特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。SYBR仅与双链DNA进行结合,因此可以通过溶解曲线,确定PCR反应是否特异。
3. 分子信标:是一种在5和3末端自身形成一个8个碱基左右的发夹结构的茎环双标记寡核苷酸探针,两端的核酸序列互补配对,导致荧光基团与淬灭基团紧紧靠近,不会产生荧光。PCR产物生成后,退火过程中,分子信标中间部分与特定DNA序列配对,荧光基因与淬灭基因分离产生荧光 。
传统定量PCR
1.传统定量PCR方法简介
1)内参照法:在不同的PCR反应管中加入已定量的内标和引物,内标用基因工程方法合成。上游引物用荧光标记,下游引物不标记。在模板扩增的同时,内标也被扩增。在PCR产物中,由于内标与靶模板的长度不同,二者的扩增产物可用电泳或高效液相分离开来,分别测定其荧光强度,以内标为对照定量待检测模板。
2)竞争法:选择由突变克隆产生的含有一个新内切位点的外源竞争性模板。在同一反应管中,待测样品与竞争模板用同一对引物同时扩增(其中一个引物为荧光标记)。扩增后用内切酶消化PCR产物,竞争性模板的产物被酶解为两个片段,而待测模板不被酶切,可通过电泳或高效液相将两种产物分开,分别测定荧光强度,根据已知模板推测未知模板的起始拷贝数。
3)PCR-ELISA法:利用地高辛或生物素等标记引物,扩增产物被固相板上特异的探针所结合,再加入抗地高辛或生物素酶标抗体-辣根过氧化物酶结合物,最终酶使底物显色。常规的PCR-ELISA法只是定性实验,若加入内标,作出标准曲线,也可实现定量检测目的。
2.内标在传统定量中的作用
由于传统定量方法都是终点检测,即PCR到达平台期后进行检测,而PCR经过对数期扩增到达平台期时,检测重现性极差。同一个模板在96孔PCR仪上做96次重复实验,所得结果有很大差异,因此无法直接从终点产物量推算出起始模板量。加入内标后,可部分消除终产物定量所造成的不准确性。但即使如此,传统的定量方法也都只能算作半定量、粗略定量的方法。
3.内标对定量PCR的影响
若在待测样品中加入已知起始拷贝数的内标,则PCR反应变为双重PCR,双重PCR反应中存在两种模板之间的干扰和竞争,尤其当两种模板的起始拷贝数相差比较大时,这种竞争会表现得更为显著。但由于待测样品的起始拷贝数是未知的,所以无法加入合适数量的已知模板作为内标。也正是这个原因,传统定量方法虽然加入内标,但仍然只是一种半定量的方法。
实时荧光定量PCR
实时荧光定量PCR技术有效地解决了传统定量只能终点检测的局限,实现了每一轮循环均检测一次荧光信号的强度,并记录在电脑软件之中,通过对每个样品Ct值的计算,根据标准曲线获得定量结果。因此,实时荧光定量PCR无需内标是建立在两个基础之上的:
1)Ct值的重现性PCR循环在到达Ct值所在的循环数时,刚刚进入真正的指数扩增期(对数期),此时微小误差尚未放大,因此Ct值的重现性极好,即同一模板不同时间扩增或同一时间不同管内扩增,得到的Ct值是恒定的。
2)Ct值与起始模板的线性关系由于Ct值与起始模板的对数存在线性关系,可利用标准曲线对未知样品进行定量测定,因此,实时荧光定量PCR是一种采用外标准曲线定量的方法。
外标准曲线的定量方法相比内标法是一种准确的、值得信赖的科学方法。利用外标准曲线的实时荧光定量PCR是迄今为止定量最准确,重现性最好的定量方法,已得到全世界的公认,广泛用于基因表达研究、转基因研究,药物疗效考核、病原体检测等诸多领域。
实时荧光定量PCR的定量方法
在Real-timePCR中,模板定量有两种策略,相对定量和绝对定量。
主词条:实时荧光定量pcr仪
临床疾病诊断
各型肝炎、艾滋病、禽流感、结核、性病等传染病诊断和疗效评价;地中海贫血、血友病、性别发育异常、智力低下综合症、胎儿畸形等优生优育检测;肿瘤标志物及瘤基因检测实现肿瘤病诊断;遗传基因检测实现遗传病诊断。
动物疾病检测
禽流感、新城疫、口蹄疫、猪瘟、沙门菌、大肠埃希菌、胸膜肺炎放线杆菌、寄生虫病等、炭疽芽孢杆菌。
食品安全
科学研究
应用行业
各级各类医疗机构、大学及研究所、CDC、检验检疫局、兽医站、食品企业及乳品厂等。
根据MIQE(Minimum Information for Publication of Quantitative Real-Time PCR Experiments) 的指导方针,qPCR为Quantitative real-time PCR的简称,RT-qPCR指的是Reverse transcription–qPCR,RT-PCR仅用来表示Reverse transcription polymerase chain reaction,而不是Real-time PCR,但是有少数作者并没有坚持这一原则。