-
X射线衍射分析 编辑
 X射线衍射分析
X射线衍射分析
发现衍射现象
 X射线衍射的产生
X射线衍射的产生
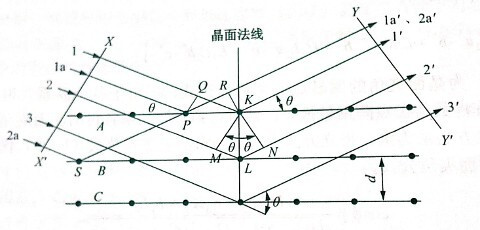
相同数量级,故由不同原子散射的X射线相互干涉,在某些特殊方向上产生强X射线衍射,衍射线在空间分布的方位和强度,与晶体结构密切相关。这就是X射线衍射的基本原理。衍射线空间方位与晶体结构的关系可用布拉格方程表示:
2dsinθ=nλ
式中:λ是X射线的波长;θ是布拉格角;d是结晶面间隔;n是整数。波长λ可用已知的X射线衍射角测定,进而求得面间隔,即结晶内原子或离子的规则排列状态。将求出的衍射X射线强度和面间隔与已知的表对照,即可确定试样结晶的物质结构,此即定性分析。从衍射X射线强度的比较,可进行定量分析。
运动学衍射理论
DArwin的理论称为X射线衍射运动学理论。该理论把衍射现象作为三维Fraunhofer衍射问题来处理,认为晶体的每个体积元的散射与其它体积元的散射无关,而且散射线通过晶体时不会再被散射。虽然这样处理可以得出足够精确的衍射方向,也能得出衍射强度,但运动学理论的根本性假设并不完全合理。因为散射线在晶体内一定会被再次散射,除了与原射线相结合外,散射线之间也能相互结合。Darwin不久以后就认识到这点,并在他的理论中作出了多重散射修正。
动力学衍射理论
Ewald的理论称为动力学理论。该理论考虑到了晶体内所有波的相互作用,认为入射线与衍射线在晶体内相干地结合,而且能来回地交换能量。两种理论对细小的晶体粉末得到的强度公式相同,而对大块完整的晶体,则必须采用动力学理论才能得出正确的结果。
发展方向
X射线分析的新发展,金属X射线分析由于设备和技术的普及已逐步变成金属研究和有机材料,纳米材料测试的常规方法。而且还用于动态测量。早期多用照相法,这种方法费时较长,强度测量的精确度低。50年代初问世的计数器衍射仪法具有快速、强度测量准确,并可配备计算机控制等优点,已经得到广泛的应用。但使用单色器的照相法在微量样品和探索未知新相的分析中仍有自己的特色。从70年代以来,随着高强度X射线源(包括超高强度的旋转阳极X射线发生器、电子同步加速辐射,高压脉冲X射线源)和高灵敏度探测器的出现以及电子计算机分析的应用,使金属 X射线学获得新的推动力。这些新技术的结合,不仅大大加快分析速度,提高精度,而且可以进行瞬时的动态观察以及对更为微弱或精细效应的研究。
原理
X射线衍射分析是利用晶体形成的X射线衍射,对物质进行内部原子在空间分布状况的结构分析方法。将具有一定波长的X射线照射到结晶性物质上时,X射线因在结晶内遇到规则排列的原子或离子而发生散射,散射的X射线在某些方向上相位得到加强,从而显示与结晶结构相对应的特有的衍射现象。衍射X射线满足布拉格(W.L.Bragg)方程:2dsinθ=nλ式中:λ是X射线的波长;θ是衍射角;d是结晶面间隔;n是整数。波长λ可用已知的X射线衍射角测定,进而求得面间隔,即结晶内原子或离子的规则排列状态。将求出的衍射X射线强度和面间隔与已知的表对照,即可确定试样结晶的物质结构,此即定性分析。从衍射X射线强度的比较,可进行定量分析。本法的特点在于可以获得元素存在的化合物状态、原子间相互结合的方式,从而可进行价态分析,可用于对环境固体污染物的物相鉴定,如大气颗粒物中的风砂和土壤成分、工业排放的金属及其化合物(粉尘)、汽车排气中卤化铅的组成、水体沉积物或悬浮物中金属存在的状态等等。
单晶衍射法
单晶X射线衍射分析的基本方法为劳埃法与周转晶体法。
劳埃法
劳埃法以光源发出连续X射线照射置于样品台上静止的单晶体样品,用平板底片记录产生的衍射线。根据底片位置的不同,劳埃法可以分为透射劳埃法和背射劳埃法。背射劳埃法不受样品厚度和吸收的限制,是常用的方法。劳埃法的衍射花样由若干劳埃斑组成,每一个劳埃斑相应于晶面的1~n级反射,各劳埃斑的分布构成一条晶带曲线。
周转晶体法
 衍射图
衍射图
多晶衍射法
多晶X射线衍射方法包括照相法与衍射仪法。
照相法
照相法以光源发出的特征X射线照射多晶样品,并用底片记录衍射花样。根据样品与底片的相对位置,照相法可以分为德拜法、聚焦法和针孔法,其中德拜法应用最为普遍。
德拜法以一束准直的特征X射线照射到小块粉末样品上,用卷成圆柱状并与样品同轴安装的窄条底片记录衍射信息,获得的衍射花样是一些衍射弧。此方法的优点为:⑴ 所用试样量少(0.1毫克即可);⑵ 包含了试样产生的全部反射线;⑶ 装置和技术比较简单。
聚焦法的底片与样品处于同一圆周上,以具有较大发散度的单色X射线照射样品上较大区域。由于同一圆周上的同弧圆周角相等,使得多晶样品中的等同晶面的衍射线在底片上聚焦成一点或一条线。聚焦法曝光时间短,分辨率是德拜法的两倍,但在小θ 范围衍射线条较少且宽,不适于分析未知样品。
针孔法用三个针孔准直的单色X射线为光源,照射到平板样品上。根据底片不同的位置针孔法又分为穿透针孔法和背射针孔法。针孔法得到的衍射花样是衍射线的整个圆环,适于研究晶粒大小、晶体完整性、宏观残余应力及多晶试样中的择优取向等。但这种方法只能记录很少的几个衍射环,不适于其它应用。
衍射仪法
X射线衍射仪以布拉格实验装置为原型,融合了机械与电子技术等多方面的成果。衍射仪由X射线发生器、X射线测角仪、辐射探测器和辐射探测电路4个基本部分组成,是以特征X射线照射多晶体样品,并以辐射探测器记录衍射信息的衍射实验装置。现代X射线衍射仪还配有控制操作和运行软件的计算机系统。X射线衍射仪的成像原理与聚集法相同,但记录方式及相应获得的衍射花样不同。衍射仪采用具有一定发散度的入射线,也用“同一圆周上的同弧圆周角相等”的原理聚焦,不同的是其聚焦圆半径随 2θ的变化而变化。衍射仪法以其方便、快捷、准确和可以自动进行数据处理等特点在许多领域中取代了照相法,现在已成为晶体结构分析等工作的主要方法。
双晶衍射法
双晶衍射仪用一束X射线(通常用Ka1作为射线源)照射一个参考晶体的表面,使符合布拉格条件的某一波长的X射线在很小角度范围内被反射,这样便得到接近单色并受到偏振化的窄反射线,再用适当的光阑作为限制,就得到近乎准值的X射线束。把此X射线作为第二晶体的入射线,第二晶体和计数管在衍射位置附近分别以Δθ 及Δ(2θ)角度摆动,就形成通常的双晶衍射仪。
在近完整晶体中,缺陷、畸变等体现在X射线谱中只有几十弧秒,而半导体材料进行外延生长要求晶格失配要达到10-4或更小。这样精细的要求使双晶X射线衍射技术成为近代光电子材料及器件研制的必备测量仪器,以双晶衍射技术为基础而发展起来的四晶及五晶衍射技术(亦称为双晶衍射),已成为近代X射线衍射技术取得突出成就的标志。但双晶衍射仪的第二晶体最好与第一晶体是同种晶体,否则会发生色散。所以在测量时,双晶衍射仪的参考晶体要与被测晶体相同,这个要求使双晶衍射仪的使用受到限制。
样品要求
1、金属样品如块状、板状、圆拄状要求磨成一个平面,面积不小于10X10毫米,如果面积太小可以用几块粘贴一起。
2、对于片状、圆拄状样品会存在严重的择优取向,衍射强度异常。因此要求测试时合 理选择响应的方向平面。
3、对于测量金属样品的微观应力(晶格畸变),测量残余奥氏体,要求样品不能简单粗磨,要求制备成金相样品,并进行普通抛光或电解抛光,消除表面应变层。
4、粉末样品要求磨成320目的粒度,约40微米。粒度粗大衍射强度低,峰形不好,分辨率低。要了解样品的物理化学性质,如是否易燃,易潮解,易腐蚀、有毒、易挥发。
5、粉末样品要求在3克左右,如果太少也需5毫克。
6、样品可以是金属、非金属、有机、无机材料粉末。
应用范围
物相分析
晶体的X射线衍射图像实质上是晶体微观结构的一种精细复杂的变换,每种晶体的结构与其X射线衍射图之间都有着一一对应的关系,其特征X射线衍射图谱不会因为它种物质混聚在一起而产生变化,这就是X射线衍射物相分析方法的依据。制备各种标准单相物质的衍射花样并使之规范化,将待分析物质的衍射花样与之对照,从而确定物质的组成相,就成为物相定性分析的基本方法。鉴定出各个相后,根据各相花样的强度正比于改组分存在的量(需要做吸收校正者除外),就可对各种组分进行定量分析。目前常用衍射仪法得到衍射图谱,用“粉末衍射标准联合会(JCPDS)”负责编辑出版的“粉末衍射卡片(PDF卡片)”进行物相分析。
目前,物相分析存在的问题主要有:⑴ 待测物图样中的最强线条可能并非某单一相的最强线,而是两个或两个以上相的某些次强或三强线叠加的结果。这时若以该线作为某相的最强线将找不到任何对应的卡片。⑵ 在众多卡片中找出满足条件的卡片,十分复杂而繁锁。虽然可以利用计算机辅助检索,但仍难以令人满意。⑶ 定量分析过程中,配制试样、绘制定标曲线或者K值测定及计算,都是复杂而艰巨的工作。为此,有人提出了可能的解决办法,认为 从相反的角度出发,根据标准数据(PDF卡片)利用计算机对定性分析的初步结果进行多相拟合显示,绘出衍射角与衍射强度的模拟衍射曲线。通过调整每一物相所占的比例,与衍射仪扫描所得的衍射图谱相比较,就可以更准确地得到定性和定量分析的结果,从而免去了一些定性分析和整个定量分析的实验和计算过程。
点阵常数的精确测定
点阵常数是晶体物质的基本结构参数,测定点阵常数在研究固态相变、确定固溶体类型、测定固溶体溶解度曲线、测定热膨胀系数等方面都得到了应用。点阵常数的测定是通过X射线衍射线的位置(θ )的测定而获得的,通过测定衍射花样中每一条衍射线的位置均可得出一个点阵常数值。
点阵常数测定中的精确度涉及两个独立的问题,即波长的精度和布拉格角的测量精度。波长的问题主要是X射线谱学家的责任,衍射工作者的任务是要在波长分布与衍射线分布之间建立一一对应的关系。知道每根反射线的密勒指数后就可以根据不同的晶系用相应的公式计算点阵常数。晶面间距测量的精度随θ 角的增加而增加, θ越大得到的点阵常数值越精确,因而点阵常数测定时应选用高角度衍射线。误差一般采用图解外推法和最小二乘法来消除,点阵常数测定的精确度极限处在1×10-5附近。
应力的测定
X射线测定应力以衍射花样特征的变化作为应变的量度。宏观应力均匀分布在物体中较大范围内,产生的均匀应变表现为该范围内方向相同的各晶粒中同名晶面间距变化相同,导致衍射线向某方向位移,这就是X射线测量宏观应力的基础;微观应力在各晶粒间甚至一个晶粒内各部分间彼此不同,产生的不均匀应变表现为某些区域晶面间距增加、某些区域晶面间距减少,结果使衍射线向不同方向位移,使其衍射线漫散宽化,这是X射线测量微观应力的基础。超微观应力在应变区内使原子偏离平衡位置,导致衍射线强度减弱,故可以通过X射线强度的变化测定超微观应力。测定应力一般用衍射仪法。
X射线测定应力具有非破坏性,可测小范围局部应力,可测表层应力,可区别应力类型、测量时无需使材料处于无应力状态等优点,但其测量精确度受组织结构的影响较大,X射线也难以测定动态瞬时应力。
晶粒尺寸和点阵畸变的测定
若多晶材料的晶粒无畸变、足够大,理论上其粉末衍射花样的谱线应特别锋利,但在实际实验中,这种谱线无法看到。这是因为仪器因素和物理因素等的综合影响,使纯衍射谱线增宽了。纯谱线的形状和宽度由试样的平均晶粒尺寸、尺寸分布以及晶体点阵中的主要缺陷决定,故对线形作适当分析,原则上可以得到上述影响因素的性质和尺度等方面的信息。
在晶粒尺寸和点阵畸变测定过程中,需要做的工作有两个:⑴ 从实验线形中得出纯衍射线形,最普遍的方法是傅里叶变换法和重复连续卷积法。⑵ 从衍射花样适当的谱线中得出晶粒尺寸和缺陷的信息。这个步骤主要是找出各种使谱线变宽的因素,并且分离这些因素对宽度的影响,从而计算出所需要的结果。主要方法有傅里叶法、线形方差法和积分宽度法。
单晶取向和多晶织构测定
单晶取向的测定就是找出晶体样品中晶体学取向与样品外坐标系的位向关系。虽然可以用光学方法等物理方法确定单晶取向,但X衍射法不仅可以精确地单晶定向,同时还能得到晶体内部微观结构的信息。一般用劳埃法单晶定向,其根据是底片上劳埃斑点转换的极射赤面投影与样品外坐标轴的极射赤面投影之间的位置关系。透射劳埃法只适用于厚度小且吸收系数小的样品;背射劳埃法就无需特别制备样品,样品厚度大小等也不受限制,因而多用此方法 。
多晶材料中晶粒取向沿一定方位偏聚的现象称为织构,常见的织构有丝织构和板织构两种类型。为反映织构的概貌和确定织构指数,有三种方法描述织构:极图、反极图和三维取向函数,这三种方法适用于不同的情况。对于丝织构,要知道其极图形式,只要求出求其丝轴指数即可,照相法和衍射仪法是可用的方法。板织构的极点分布比较复杂,需要两个指数来表示,且多用衍射仪进行测定。