-
度洛西汀 编辑
度洛西汀是一种选择性的5-羟色胺(5-HT)和去甲肾上腺素(NE)再摄取抑制药。临床前研究结果显示,度洛西汀是神经元5-HT与NE再摄取的强抑制剂,对多巴胺再摄取的抑制作用相对较弱。体外研究结果显示,度洛西汀与多巴胺能受体、肾上腺素受体、胆碱能受体、组胺能受体、阿片受体、谷氨酸受体、GABA受体无明显亲和力。度洛西汀不抑制单胺氧化酶。
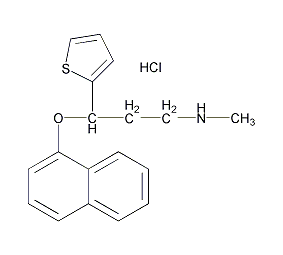
中文别名:(R)-(-)-N-甲基-3-(1-萘氧基)-3-(2-噻吩)-丙胺;右旋度洛西汀盐酸盐
英文名称:(3R)-N-methyl-3-naphthalen-1-yloxy-3-thiophen-2-ylpropan-1-amine
英文别名:Ariclaim; N-methyl-3-napthalen-1-oxy-3-thiophen-2-yl-1-amine; R-Duloxetine HCl; UNII-TK9VOT90JQ; Cymbalta
CAS号:116539-60-7
分子量:333.87600
精确质量:333.09500
PSA:49.50000
LogP:5.82380
结构式:
物化性质
密度:1.158g/cm3
沸点:466.2ºC at 760 mmHg
闪点:235.7ºC
分子结构数据
2、摩尔体积(cm3/mol):256.8
3、等张比容(90.2K):669.0
4、表面张力(dyne/cm):46.0
5、极化率(10-24cm3):36.13
计算化学数据
2.氢键供体数量:1
3.氢键受体数量:3
4.可旋转化学键数量:6
5.互变异构体数量:无
6.拓扑分子极性表面积49.5
7.重原子数量:21
8.表面电荷:0
9.复杂度:312
10.同位素原子数量:0
11.确定原子立构中心数量:1
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:0
14.不确定化学键立构中心数量:0
15.共价键单元数量:1
性质与稳定性
盐酸度洛西汀(Duloxetine Hydrochloride):C18H19NOS·HCI。。白色固体。二甲基甲酰胺-水(66:34)的pKa为9.6 。
合成方法
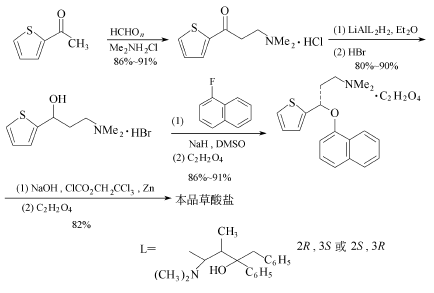
下列噻吩化合物经Marmieh反应,然后用Yarnaguchi-Masher-Pohland(YMP)复合试剂LiAl2H2在乙醚中,于-78℃进行不对称还原,而后加入氢溴酸,使还原产物以氢溴酸盐形式得到。再和1-氟萘进行烷基化反应,最后去甲基化,产物以草酸盐的形式得到。
药物剂型
1.度洛西汀胶囊:20mg,30mg,40mg,60mg。贮法:室温保存。2.度洛西汀肠溶片。3.度洛西汀肠溶胶囊。
药理作用
度洛西汀是一种选择性的5-羟色胺(5-HT)和去甲肾上腺素(NE)再摄取抑制药。度洛西汀抗抑郁与中枢镇痛作用的机制尚未明确,但认为与其增强中枢神经系统5-羟色胺能与去甲肾上腺素能功能有关。临床前研究结果显示,度洛西汀是神经元5-HT与NE再摄取的强抑制剂,对多巴胺再摄取的抑制作用相对较弱。体外研究结果显示,度洛西汀与多巴胺能受体、肾上腺素受体、胆碱能受体、组胺能受体、阿片受体、谷氨酸受体、GABA受体无明显亲和力。度洛西汀不抑制单胺氧化酶。
药动学
本药口服治疗抑郁症3周内起效,达峰时间为4~6h,多剂量给药作用可持续7天以上。血药浓度达峰时间为6~10h。本药口服生物利用度高于70%,总蛋白结合率高于95%,表观分布容积为1640L。在肝脏代谢,代谢产物为去甲基度洛西汀、羟化代谢产物。本药肾脏排泄率为77%,主要以代谢产物的形式排出;15%随粪便排泄。总体清除率为114L/h,原形药消除半衰期为11~16h。
适应证
1.用于治疗重型抑郁症。2.用于糖尿病周围神经痛。3.用于女性中至重度应激性尿失禁。
禁忌证
1.对本药过敏者。2.未控制的窄角型青光眼患者。3.不推荐肝功能不全者使用。
注意事项
1.慎用:(1)经控制的窄角型青光眼患者。(2)癫痫患者。(3)躁狂或轻躁狂活动期患者。(4)有自杀倾向的抑郁症患者。(5)肾功能不全者。2.药物对妊娠的影响:妊娠期使用本药,可使新生儿发生严重并发症(呼吸窘迫、窒息、发绀、癫痫发作、体温不稳定、呕吐、低血糖、肌张力下降或升高、反射亢进、神经过敏性震颤及易激惹等)。美国药品和食品管理局(FDA)对本药的妊娠安全性分级为C级。3.药物对哺乳的影响:尚不明确。4.本药禁止与5-羟色胺能药物合用。5.如出现血压持续上升,应予密切监测。6.停药应逐渐减量,突然撤药可出现撤药综合征。
不良反应
1.心血管系统:可引起血压轻度上升及心率下降,甚至血压持续上升。2.中枢神经系统:可见失眠、头痛、嗜睡、晕眩、震颤及易激惹。3.代谢/内分泌系统:可见体重下降。4.泌尿生殖系统:可见排尿困难及男性性功能障碍(如射精障碍、性欲下降、勃起障碍、射精延迟、达高潮能力障碍)。5.胃肠道:可见恶心、腹泻、便秘、口干、纳差及味觉改变。6.血液:较少见贫血、白细胞减少、白细胞计数升高、淋巴结病及血小板减少。7.皮肤:常见盗汗、瘙痒及皮疹。较少见痤疮、脱发、冷汗、瘀斑、湿疹、红斑、颜面部水肿及光敏反应。另可见出汗增多(6%)。8.眼:可见视物模糊(4%)。
用法用量
成人:1.常规剂量 口服给药:(1)抑郁症:①1次20~30mg,2次/d。②1天60mg,顿服。(2)糖尿病神经痛:1天60mg,顿服。对可能出现耐受的患者可降低起始剂量。(3)女性中至重度应激性尿失禁:起始剂量1次40mg,2次/d,如不能耐受,则4周后减量至1次20mg,2次/d。2.肾功能不全时剂量 肾功能不全时应使用较低的起始剂量,逐渐增量。不推荐终末期肾病(需要透析)或严重肾功能损害(肌酐清除率小于30ml/min)患者使用。
药物相应作用
1.本药与单胺氧化酶抑制药(MAOIs)均抑制5-HT代谢,两药合用易出现严重不良反应,如中枢神经毒性或5-HT综合征(其临床表现为高血压、高热、肌阵挛、激惹及烦躁不安、反射亢进、出汗、寒战及震颤),甚至死亡。禁止本药与MAOIs合用;停用MAOIs 14天后才能使用本药;停用本药5天后才能使用MAOIs。2.卷曲霉素、依诺沙星、氟伏沙明及奎尼丁可抑制本药的代谢,增加本药血药浓度(或生物利用度)及毒性,两者合用须监测不良反应,需要时减少本药剂量。3.本药与氟西汀、帕罗西汀合用,互相抑制代谢,两药的生物利用度、血药浓度均增加,发生严重不良反应的危险性增加,合用时应调整两药剂量。4.本药可抑制三环类抗抑郁药(如阿米替林)的代谢,两者合用,本药可增加后者的生物利用度、血药浓度及毒性。如必须合用,应密切监测三环类抗抑郁药的血药浓度、中毒的症状及体征(抗胆碱能作用、过度镇静、意识混乱及心律失常)。5.本药可抑制吩噻嗪类药(奋乃静)的代谢,增加后者的血药浓度及毒性(过度镇静、意识障碍、心律失常、直立性低血压、高热及锥体外系反应)。两者合用应监测不良反应,必要时减少剂量。6.本药可抑制硫利达嗪的代谢,增加后者血药浓度及心脏毒性(QT间期延长、尖端扭转性室性心动过速、心脏停搏),两者不应合用。7.本药可抑制Ic类抗心律失常药的代谢,增加后者的血药浓度及心脏毒性。两者合用应密切监测Ic类抗心律失常药的血药浓度及心电图。8.本药与中枢神经系统抑制药合用,可引起精神运动性障碍恶化,禁止两者合用。食物不影响本药的血药峰浓度,但可减慢吸收,并降低吸收度10% 。








