-
northern印迹杂交 编辑
中文名:northern印迹杂交
外文名:Northernblot
pH:高于4.0
关联技术:Westernblot
Northern印迹杂交的RNA吸印与Southern印迹杂交的DNA吸印方法类似,只是在上样前用甲基氢氧化银、乙二醛或甲醛使RNA变性,而不用NaOH,因为它会水解RNA的2'-羟基基团。RNA变性后有利于在转印过程中与硝酸纤维素膜结合,它同样可在高盐中进行转印,但在烘烤前与膜结合得并不牢固,所以在转印后用低盐缓冲液洗脱,否则RNA会被洗脱。在胶中不能加EB,因为它会影响RNA与硝酸纤维素膜的结合。为测定片段大小,可在同一块胶上加分子量标记物一同电泳,之后将标记物切下、上色、照相,样品胶则进行Northern转印。标记物胶上色的方法是在暗室中将其浸在含5μg/ml EB的0.1mol/L醋酸铵中10min,光在水中就可脱色,在紫外光下用一次成像相机拍照时,上色的RNA胶要尽可能少接触紫外光,若接触太多或在白炽灯下暴露过久,会使RNA信号降低。
琼脂糖凝胶中分离功能完整的mRNA时,甲基氢氧化银是一种强力、可逆变性剂,但是有毒,因而许多人喜用甲醛作为变性剂。所有操作均应避免RNase的污染。
原理:
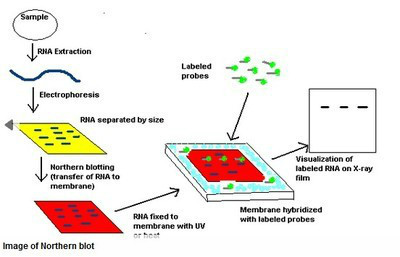
整合到植物染色体上的外源基因如果能正常表达,则转化植株细胞内有其转录产物——特异mRNA的生成。将提取的植物总RNA或mRNA用变性凝胶电泳分离,则不同的RNA分子将按分子质量大小依次排布在凝胶上;将他们原位转移到固定膜上;在适宜的离子强度及温度条件下,用探针与膜杂交;然后通过探针的标记性质检测出杂交体。若经杂交,样品无杂交带出现,表明外源基因已经整合到植物细胞染色体上,但在该取材部位及生理状态下该基因并未有效表达。
试剂
10×MSE缓冲液:0.2mol/L吗啉代丙烷磺酸(MOPS),pH7.0,50mmol/L醋酸钠,1mmol/LEDTApH8.0。
5×载样缓冲液:50%甘油,1mmol/LEDTA,0.4%溴酚蓝。
甲醛:用水配成37%浓度(12.3mol/L),应在通风柜中操作,pH高于4.0。
20×SSC;
去离子甲酰胺;
50mmol/LNaOH(含10 mmol/L NaCl);
0.1mol/LTris,pH7.5。
步骤
40ml水中加7g琼脂糖,煮沸溶解,冷却到60℃,加7 ml 10×MSE缓冲液,11.5ml甲醛,加水定容至70ml,混匀后倒入盛胶槽。
等胶凝固后,去掉梳子和胶布,将盛胶槽放入1×MSE缓冲液的电泳槽。
使RNA变性(最多20μg),RNA4.5ml,10×MSE缓冲液20ml,甲醛3.5ml,去离子甲酰胺10ml。
55℃加热15min,冰浴冷却。
加2 ml 5×载样缓冲液。
60伏电泳过夜。
取出凝胶,水中浸泡2次,每次5min。
室温下将胶浸到50mmol/LNaOH和10mmol/LNaCl中45min,水解高分子RNA,以增强转印。
室温下将胶浸到0.1mmol/L TriShcl(pH7.5)中45min,使胶中和。
20×SSC洗胶1h。
20×SSC中过夜转印到硝酸纤维素膜上。
取出硝酸纤维素膜,80℃真空烘烤2h。
注意事项
严格遵守试验规则,务必准确。
由于好多药品是有毒的,对人体有害,请注意自身安全,做好防护。
①Northern杂交后继过程的试剂能不能不用DEPC处理?不处理会不会影响结果? 做Northern blot时,一般要求是所有的试剂都必须用DEPC处理,这是操作流程的要求,以抑制RNA酶的活性。DEPC是一种致癌剂,须小心操作,尤其在用DEPC处理乙酸铵时,因为DEPC能与铵离子反应产生氨基甲酸乙酯(一种潜在的致癌剂)。也有人认为杂交液、预杂交液、洗膜液实际上可以不用DEPC处理,但杂交液(无探针)、预杂交液、洗膜液必须要在15磅的条件下高压30min,这样做Northern blot也可以做出来,否则就有可能失败。 |
②DEPC处理试剂会不会对所配置的溶液的pH值造成影响? DEPC是极弱酸性的,由于其处理水的浓度仅有万分之一,故用DEPC处理试剂不会导致其pH值明显变化。 |
③以Northern杂交鉴定RNA在组织中的表达量,判断依据是否就是最后膜条带显影的亮度? 一般可以认为Northern杂交是mRNA绝对定量的方法,但必须参考内参的信号,才能估算不同组织中的表达量。 |
④背景很高 背景很高的原因有多种: ·标记探针时,未掺入的dNTPs没有完全从探针中除去。建议使用PCR产物纯化柱来纯化探针。 ·DNA探针的平均大小过大。最佳大小范围为200~800核苷酸。 ·杂交溶液中探针浓度过高。对于DNA探针来说,浓度不要超过10ng/ml;对于寡聚核苷酸探针来说,不要超过50ng/ml。 ·封闭不彻底。 ·不管是在何种条件下洗涤,如果延长洗膜时间仍不能改善背景状况的话,则要洗脱膜上的探针重新进行杂交。 |
⑤没有杂交信号或杂交信号很弱 可能有以下原因: ·RNA降解。解决方案参见①。 ·上样量不足。一定要保证足够的上样量,尤其是在检测低丰度RNA时,总RNA通常需要50μg,溶于上样缓冲液中。 ·电泳条件不合适。比如胶浓度太小、电泳时加样孔偏长或电泳时间偏长,均易使较小的RNA片断弥散,电泳0.5~1h为宜。 ·如果X线胶片曝光30~60min后仍没有杂交信号产生,可能是杂交探针的活性很低,需重新标记探针。必要时,可采用Sephades G-50柱层析法纯化标记的探针,以去除标记反应中未结合的(游离的)核苷酸。 ·如果重新标记后探针的比活性仍然很低,则可能是标记时DNA的用量太少。DNA的用量通常为25~50ng(可根据浓的母液的OD260值计算)。如果手头上DNA的样品量很少,可将标记时使用的DNA量点样在琼脂糖凝胶上,旁边点上已知含量的DNA标准样品一起电泳,从中估算出探针DNA的量。经溴化乙锭染色后,如果看不到明显的探针DNA带,说明DNA量少于20~50ng,那么在标记时要加大2~3倍的DNA量如果仍然失败,则要用已知含量的对照探针(如β-actin探针)来优化标记条件。 ·还有一种可能是探针与靶基因不完全同源。如果是用种间交叉的探针进行杂交,则应降低最后洗涤条件的严格程度,将洗涤温度提高到50~56℃,并用洗涤液1代替洗涤液3。如果是用合成的寡聚核苷酸探针进行杂交,则应确保探针与靶基因完全同源。 |
⑥无法去除探针和再次杂交 杂交膜如果不能再次杂交,很可能是上次杂交的探针没有完全从膜上去除,也可能是因膜已经干燥或部分干燥造成的。如果膜已经干燥哪怕只是部分干燥,要想除去膜上的探针已是不可能的了。因此,在整个洗膜过程中应避免使膜干燥。在完成洗涤后,用镊子将膜夹起滴去多余的洗涤液(不要使膜干燥),立即用塑料薄膜将膜包裹住。 |
⑦杂交2次后信号减弱 这种现象在低丰度基因中极为常见,即使是高丰度基因(如β-actin),经过2轮杂交后,杂交信号也不可能像通常看到的那样强。 |
⑧杂交膜能否重复使用? 结合了待测RNA的膜与探针杂交后,可经碱或热变性方法将探针洗脱,膜可反复使用与其他探针杂交。方法如下:杂交的膜(注意:杂交过的膜在保存过程中不能干燥,否则探针将会与膜形成不可逆的结合)置100℃ 0.5% SDS中煮沸3min,自然冷却至室温后,将膜放入双蒸水中漂洗2~3遍。取出膜,用滤纸吸去膜表面的水分。将膜直接进行另一种探针的杂交或用保鲜膜包好,室温下真空保存。尼龙膜可反复使用5次以上。 |
⑨其他 |