-
共价键 编辑
早期历史
 图1
图1
随后,原子论者德谟克利特设想,原子与原子间,存在着一种“钩子”,也可以说是粗糙的表面,以致它们在相互碰撞时黏在一起,构成了一个稳定的聚集体。德谟克利特对化学键的设想相比于之前的自然哲学家,是更加先进的,他剔除了此类设想中的唯心主义因素。
中世纪的J.R.格劳伯则提出了物质同类相亲、异类相斥的思想。其后还出现了关于物质结合的亲和力说,认为物质的微粒具有亲和力,由此互相吸引而结合在一起。总之,人们关于化学键朦胧的认识,启发了后来的化学家。
近代史
18世纪,燃素(phlogiston)的概念进入了化学,并为恩斯特·施塔尔(Ernst Stahl)、亨利·卡文迪许(Henry Cavendish)和约瑟夫·普利斯特里(Joseph Priestley)等先进的化学家所接受。当时,牛顿力学已经提出,他们希望把原子间的作用力和牛顿力学结合起来,给出经典物理学的解释,但限于当时的条件,这无疑是无法完成的。
1916年,德国化学家沃尔特·柯塞尔(Walther Kossel)在考察大量事实后得出结论:任何元素的原子都要使最外层满足8电子稳定结构,但科塞尔只解释了离子化合物的形成过程,并没有解释共价键的形成。
1919年,化学家欧文·朗缪尔首次使用“共价”来描述原子间的成键过程 “(原文)we shall deNOte by the termcovalencethe number of pairs of eleCTrons which a given atom shares with its neighbors ”(我们应该用“共价”一词表示原子间通过共用电子对形成的作用力)。
1922年,尼尔斯·玻尔(N.Bohr)从量子化的角度重新审视了卢瑟福的核式模型,这为化学家对化学键的认识,提供了全新的平台,他认为电子应该位于确定的轨道之中,并且能够在不同轨道之间跃迁,定态跃迁可以很好的解释氢原子光谱的各个谱线。
 图2
图2
1924年,路易斯·德布罗意(Louis de Broglie)提出波粒二象性的假说,建立了一个原子的数学模型,用来将电子描述为一个三维波形。在数学上不能够同时得到位置和动量的精确值。
1926年,薛定谔提出量子力学的波动方程,它可以直接用来解释化学键的“形成”和“断裂”,这成为量子化学最初的开端。
1927年,沃尔特·海特勒(W.H.Heitler)和弗里茨·伦敦(F.London)用量子力学处理氢分子,用近似方法算出了氢分子体系的波函数,首次用量子力学方法解决共价键问题。价键理论在这一方法的推广中诞生,他们研究共价键的方法就被称为HL法。
1928年,恩利克·费米(Enrica Fermi)提出了一个基于泊松分布的单电子密度模型试图解决原子结构问题。 之后,道格拉斯·哈特里(Douglas Rayner Hartree)运用迭代法,将体系电子的哈密顿算子分解为若干个单电子哈密顿算子的简单加和,进而将体系多电子波函数表示为单电子波函数的积,改进这一模型,提出哈特里方程。
1930年,哈特里的学生福克(Fock)与约翰·斯莱特(John Clarke Slater)完善了哈特里方程,称为哈特里-福克方程(HF)。50年代初,斯莱特得到了HF的近似波函数:哈特里-福克-斯莱特方程(HFS) 。1963年,赫尔曼(F.Hermann)和斯基尔曼(S.Skillman)把HFS应用于基态原子函数。
1950年,克莱蒙斯·罗瑟恩(C. C. J. Roothaan)进一步提出将方程中的分子轨道用组成分子的原子轨道线性展开,发展出了著名的RHF方程,1964年,计算机化学家恩里克·克莱门蒂(E.Clementi)发表了大量的RHF波函数, 该方程以及后续的改进版已经成为现代处理量子化学问题的主要方法。
 图3
图3
1932年,弗里德里希·洪德(F.Hund)将共价键分为σ键、π键、δ键三种,使价键理论进一步系统化,与经典的化合价理论有机地结合起来。
同年,美国化学家罗伯特·马利肯(RobERT S.Mulliken)提出分子轨道理论。认为化合物中的电子不属于某个原子,而是在整个分子内运动。他的方法和经典化学相距太远,计算又很繁琐,一时不被化学界所接受。后经过罗伯特·密立根(Robert A.Millikan)、菲利普·伦纳德(Philipp Lenard)、埃里希·休克尔(Erich Hückel)等人的完善,在化学界逐渐得到认可。
1940年,亨利·希吉维克(H.Sidgwick)和托马斯·坡维尔(Thomas A.Powell)在总结实验事实的基础上提出了一种简单的理论模型,用以预测简单分子或离子的立体结构。这种理论模型后经罗纳德·吉列斯比(R.J.Gillespie)和罗纳德·尼霍尔姆(R.S.Nyholm)在20世纪50年代加以发展,定名为价层电子对互斥理论,简称VSEPR。VSEPR与轨道杂化理论相结合,可以半定量地推测分子的成键方式与分子结构。
1951年,福井谦一提出前线轨道理论,认为,分子中能量最高的分子轨道(HOMO)和没有被电子占据的,能量最低的分子轨道(LUMO)是决定一个体系发生化学反应的关键,其他能量的分子轨道对于化学反应虽然有影响但是影响很小,可以暂时忽略。HOMO和LUMO便是所谓前线轨道。
1965年,美国化学家罗伯·伍德沃德(Rober B.Woodward)与霍夫曼参照福井谦一的前线轨道理论,提出了分子轨道对称守恒原理。分子轨道理论得到了新的发展。
由于计算机技术的迅猛发展,和蒙特卡罗方法的应用,量子化学与计算机化学日新月异,对分子结构的推算变得愈发精确期间也诞生了一大批优秀的化学家,据估计,21世纪中期,量子化学还将有新的突破。
饱和性
在共价键的形成过程中,因为每个原子所能提供的未成对电子数是一定的,一个原子的一个未成对电子与其他原子的未成对电子配对后,就不能再与其它电子配对,即,每个原子能形成的共价键总数是一定的,这就是共价键的饱和性。
共价键的饱和性决定了各种原子形成分子时相互结合的数量关系 ,是定比定律(law of definite proportion)的内在原因之一。
方向性
除s轨道是球形的以外,其它原子轨道都有其固定的延展方向,所以共价键在形成时,轨道重叠也有固定的方向,共价键也有它的方向性,共价键的方向决定着分子的构形。
影响共价键的方向性的因素为轨道伸展方向。
化学变化的本质是旧键的断裂和新键的形成,化学反应中,共价键存在两种断裂方式,在化学反应尤其是有机化学中有重要影响。
均裂与自由基反应
共价键在发生均裂时,成键电子平均分给两个原子(团),均裂产生的带单电子的原子(团)称为自由基,用“R·”表示,自由基具有反应活性,能参与化学反应,自由基反应一般在光或热的作用下进行。
异裂与离子型反应
共价键发生异裂时生成正、负离子,例如氯化氢在水中电离成氢离子和氯离子。有机物共价键异裂生成的碳正离子和负离子是有机反应的活泼物种,往往在生成的一瞬间就参加反应,但可以证明其存在。
由共价键异裂引发的反应称离子型反应,其下又可分为两种
·亲电反应(elECTrophilic reaction)
·亲核反应(nucleophilic reaction)
离子型反应一般在酸碱或极性物质的催化下进行。
路易斯理论
路易斯理论,又称“八隅体规则”、“电子配对理论”是最早提出的,具有划时代意义的共价键理论,它没有量子力学基础,但因为简单易懂,也能解释大部分共价键的形成,至今依然出现在中学课本里。
共用电子对理论有以下几点:
1、原子最外层达到8电子时是稳定结构,化合物中的所有原子的最外层价电子数必须为8(氢为2);
2、原子间形成共价键时,可通过共用电子的方式使最外层达到8(2)电子稳定结构。
路易斯理论的电子配对思想为价键理论的发展奠定了基础。 值得注意的是,路易斯理论尚不完善,它无法说明电子配对的原因和实质;此外,不符合“八隅体规则”的化合物也有很多,例如:三氟化硼(6电子)、五氯化磷(10电子)、六氟化硫(12电子)。
价键理论
价键理论是基于路易斯理论电子配对思想发展起来的共价键理论。价键理论将应用量子力学解决氢分子问题的成果推广到其他共价化合物中,成功解释了许多分子的结构问题。
海特勒-伦敦法
沃尔特·海特勒(W.H.Heitler)和弗里茨·伦敦(F.London)在运用量子力学方法处理氢气分子的过程中,得到了分子能量E和核间距R之间的关系曲线,发现:若两个氢原子自旋方向相反,随着轨道的重叠(波函数相加)会出现一个概率密度较大的区域,氢原子将在系统能量最低核间距处成键;若两个氢原子自旋方向相同,则相减的波函数单调递减,系统能量无限趋近E=0,没有最低点,无法成键。因此,价键理论通过对氢分子的研究阐明了电子配对的内在原因和共价键的本质,价键理论就在HL的推广中诞生。
轨道杂化理论
价键理论在解释分子中各原子分布情况时,莱纳斯·鲍林(L.Pauling)提出了轨道杂化理论。理论要点有
1、中心原子能量相近的不同轨道在外界的影响下会发生杂化,形成新的轨道,称杂化原子轨道,简称杂化轨道;
2、杂化轨道在角度分布上,比单纯的原子轨道更为集中,因而重叠程度也更大,更加利于成键;
3、参加杂化的原子轨道数目与形成的杂化轨道数目相等,不同类型的杂化轨道,其空间取向不同。
杂化类型 | 杂化轨道夹角 | 空间取向 |
|---|---|---|
sp^1 | 180 | 直线型 |
sp^2 | 120 | 平面正三角形 |
sp^3 | 109.28 | 正四面体 |
sp^3d (dsp^3) | 90 120 | 三角双锥 |
sp^3d^2 (d^2sp^3) | 90 | 正八面体 |
注:此为杂化轨道的空间取向,不是化合物的结构
在化合物中,这些轨道可能被孤对电子或单电子填充,例如,N原子进行sp²杂化形成的NO2分子中,有一个单电子,NO2的空间结构是折线形(正三角形的一个顶点是单电子,电子是“看不见”的)。
互斥理论
价层电子对互斥理论(VSEPR Theory)是一个用来预测单个共价分子形态的化学模型。理论通过计算中心原子的价层电子数和配位数来预测分子的几何构型,其理论要点有:
1、共价分子中,中心原子周围电子对排布的几何形状,主要决定于中心原子的价电子层中的电子对数(包括成键电子对和孤对电子)。这些电子的位置倾向于分离的尽可能远一些,使彼此受到的排斥力最小 ;
2、电子层中电子对相互排斥作用的大小,取决于电子对间的相互角度和电子对的成键情况。相距角度小,排斥力大。成键电子对因受两个原子吸引,电子云较为紧缩,对其相邻电子对的斥力小于仅受一个原子核吸引的孤对电子对其相邻电子对的斥力。即,电子对间斥力大小顺序为:孤对电子-孤对电子>孤对电子-成键电子对>成键电子对-成键电子对;
3、分子中的双键、三键当作单键处理;
推测分子构形
设中心原子为A,其余n个配位原子均用B表示,m对孤对电子用E表示,则该物质可表示为ABnEm。令z=n+m,B和E都用Y表示,则该物质可表示为AYz,这里的Y就表示中心原子的价电子层中的电子对,z就表示中心原子的价电子层中的电子对数。我们可根据如下公式推测分子构型:
n由化学式即可看出
m=1/2(中心原子价电子数-配位原子提供的电子总数±离子电荷数)
z=n+m
Z=n+m | 2 | 3 | 4 | 5 | 6 |
结构形式 | 直线型 | 平面三角形 | 四面体 | 三角双锥 | 八面体 |
注:更详细的表参见wikipedia,VSEPR theory(扩展阅读)
分子轨道理论
分子轨道理论是比价键理论更精确的方法,其理论要点有
1、分子中的电子不属于某个原子轨道,而属于整个分子;
2、分子轨道由原子轨道线性组合而成,分子轨道数目等于组成分子轨道的原子轨道数目,其中些轨道能量降低,成为“成键轨道”另一些能量升高,成为“反键轨道”,还有一些能量不变,称“非键轨道”;
3、原子轨道在线性组合时,遵守“对称性匹配原则”、“能量相近原则”、“最大重叠原则”;
4、电子在分子轨道中排布时,遵守“能量最低原理”、“泡利不相容原理”、“洪特规则”;
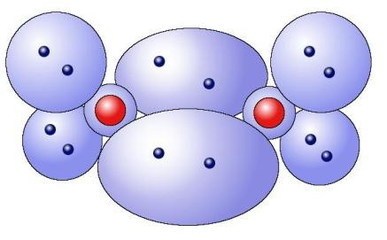
分子轨道理论能解释一些价键理论无法解释的现象,比如氧分子的顺磁性。
氧原子的外层电子数为6,这六个电子中的四个组成两对,其它两个单独存在。
 图4 每个氧原子有六个外层电子
图4 每个氧原子有六个外层电子
这两个单独的电子与另一个原子中相应的单独的电子结合组成两个新的共用的电子对,由此达到电子饱和的状态。
 图5 氧分子O2的模型
图5 氧分子O2的模型
共价键从不同的角度可以进行不同的分类,每一种分类都包括了所有的共价键(只是分类角度不同)。
按成键方式
 图6 σ键
图6 σ键
由两个原子轨道沿轨道对称轴方向相互重叠导致电子在核间出现概率增大而形成的共价键,叫做σ键,可以简记为“头碰头”(见图6)。 σ键属于定域键,它可以是一般共价键,也可以是配位共价键。一般的单键都是σ键。原子轨道发生杂化后形成的共价键也是σ键。由于σ键是沿轨道对称轴方向形成的,轨道间重叠程度大,所以,通常σ键的键能比较大,不易断裂,而且,由于有效重叠只有一次,所以两个原子间至多只能形成一条σ键。
π键(pi bond)
 图7 π键
图7 π键
 图8 苯分子中的大π键
图8 苯分子中的大π键
δ键(delta bond)
 图9 δ键
图9 δ键
δ键只有两个节面(电子云密度为零的平面)。从键轴看去,δ键的轨道对称性与d轨道的没有区别,而希腊字母δ也正来源于d轨道。
δ键常出现在有机金属化合物中,尤其是钌、钼和铼所形成的化合物。通常所说的“四重键”指的就是一个σ键、两个π键和一个δ键。
以上三种化学键经过组合,可以形成各种不同的键型,例如,一个σ键和两个π键可以组成一个三键,但有证据表明双原子间的共价键最多不能超过六条。
共价键是电子云的重叠,所以共价键最本质的分类方式就是它们的重叠方式。现在已知有3种重叠方式,分别称作:
σ键π键δ键在有机化合物中,通常把共价键以其共用的电子对数分为单键、双键以及三键。单键是一根σ键;双键和三键都含一根σ键,其余1根或2根是π键。
但无机化合物不用此法。原因是,无机化合物中经常出现的共轭体系(离域π键)使得某两个原子之间共用的电子对数很难确定,因此无机物中常取平均键级,作为键能的粗略标准。
按成键过程
1、一般共价键
一般共价键有时也称“正常共价键”,是为了和“配位共价键”进行区分时使用的概念,指成键时两个原子各自提供一个未成对电子形成的共价键。
2、配位共价键(coordinate covalent bond)
配位共价键简称“配位键”是指两原子的成键电子全部由一个原子提供所形成的共价键,其中,提供所有成键电子的称“配位体(简称配体)”、提供空轨道接纳电子的称“受体”。常见的配体有:氨气(氮原子)、一氧化碳(碳原子)、氰根离子(碳原子)、水(氧原子)、氢氧根(氧原子);受体是多种多样的:有氢离子、以三氟化硼(硼原子)为代表的缺电子化合物、还有大量过渡金属元素。对配位化合物的研究已经发展为一门专门的学科,配位化学。
配位键是一种特殊的共价键,它的特点在于共用的一对电子出自同一原子。形成配位键的条件是,一个原子有孤电子对,而另一个原子有空轨道。
中心离子:在配合物中,提供空轨道的一方称为中心离子
配体:在配合物中,提供孤对电子的一方称为配体
分类 | 化学键 |
|---|---|
共价键 | |
π键:反馈π键·共轭·超共轭效应·方向性 | |
δ键:四重键·五重键·六重键 | |
氢键 | |
范德华力·机械结合作用·嵌入·卤键·亲金作用·重叠·熵力·极性 | |
其他 |
2.1配位共价键与一般共价键的异同
配位共价键与一般共价键的区别只体现在成键过程上,它们的键参数是相同的,例如,铵根离子的氮氢键中,有三条是一般共价键,一条是配位共价键,但这四条键完全等价,铵根离子也是完全对称的正四面体形。在书写时,一般共价键使用符号“—”;配位共价键使用符号“→”箭头从配体指向受体。
电子偏向
1、极性共价键(polar bond)
 图10 极性键,标&
图10 极性键,标&
2、非极性共价键(non-polar bond)
由同种元素的原子间形成的共价键,叫做非极性共价键。同种原子吸引共用电子对的能力相等,成键电子对匀称地分布在两核之间,不偏向任何一个原子,成键的原子都不显电性。 非极性共价键存在于单质中,也存在于某些化合物中,完全由非极性键构成的分子一定是非极性分子(但有的非极性分子中含有极性键)。
键参数
1、键长(bond length)
键长指两个成键原子的平衡核间距离,是了解分子结构的基本构型参数,也是了解化学键强弱和性质的参数。 对于由相同的A和B两个原子组成的化学键,键长值小,键强; 键的数目多,键长值小。 在实际的分子中,由于受共轭效应、空间阻碍效应和相邻基团电负性的影响,同一种化学键键长还有一定差异。 键长的测定主要是通过分子光谱和热化学手段。 下表给出常见共价键的键长(pm)数据取自《化学-物质结构与性质(选修)》(2007年)。
共价键 | 键长 | 共价键 | 键长 | 共价键 | 键长 | 共价键 | 键长 |
|---|---|---|---|---|---|---|---|
H-H | 74 | H-F | 92 | H-Cl | 127 | H-Br | 141 |
H-I | 161 | C-H | 109 | C-C | 154 | C-Si | 186 |
C-N | 147 | C-O | 143 | C-P | 187 | C-S | 181 |
C-F | 133 | C-Cl | 177 | C-Br | 194 | C-I | 213 |
N-H | 101 | N-N | 146 | N-P | 177 | N-O | 144 |
N-F | 139 | N-Cl | 191 | N-Br | 214 | N-I | 222 |
O-H | 101 | O-P | 160 | O-S | 151 | O-F | 142 |
O-Cl | 164 | O-Br | 172 | O-I | 194 | Si-H | 148 |
Si-Si | 234 | Si-O | 161 | SI-S | 210 | Si-F | 156 |
Si-Cl | 204 | Si-Br | 216 | Si-I | 240 | P-H | 142 |
P-Si | 227 | P-P | 221 | P-F | 156 | P-Br | 222 |
P-I | 243 | S-H | 134 | S-F | 158 | S-Cl | 201 |
S-Br | 225 | S-I | 234 | F-F | 143 | Cl-Cl | 199 |
Br-Br | 228 | I-I | 266 | C=C | 134 | C=N | 127 |
C=O | 123 | N=N | 122 | N=O | 120 | O=O | 121 |
C≡C | 121 | C≡N | 115 | N≡N | 110 |
2、键能(bond energy)
通常指在标准状态下气态分子拆开成气态原子时,每种键所需能量的平均值。对双原子分子来说,键能就是键的解离能。键能与键焓近似相等,气态分子的原子化能等于全部键能之和。
下表给出常见共价键的键能(kJ/mol)数据取自《化学-物质结构与性质(选修)》(2007年)。
共价键 | 键能 | 共价键 | 键能 | 共价键 | 键能 | 共价键 | 键能 |
|---|---|---|---|---|---|---|---|
H-H | 436 | H-F | 565 | H-Cl | 431 | H-Br | 363 |
H-I | 297 | C-H | 413 | C-C | 347 | C-Si | 301 |
C-N | 305 | C-O | 358 | C-P | 264 | C-S | 259 |
C-F | 453 | C-Cl | 339 | C-Br | 276 | C-I | 216 |
N-H | 391 | N-N | 160 | N-P | 209 | N-O | 201 |
N-F | 272 | N-Cl | 200 | N-Br | 243 | N-I | 159 |
O-H | 467 | O-P | 351 | O-S | 265 | O-F | 190 |
O-Cl | 203 | O-Br | 237.6 | O-I | 不详 | Si-H | 323 |
Si-Si | 226 | Si-O | 368 | Si-S | 226 | Si-F | 565 |
Si-Cl | 381 | Si-Br | 213 | Si-I | 不详 | P-H | 320 |
P-Si | 310 | P-P | 200 | P-F | 490 | P-Br | 272 |
P-I | 184 | S-H | 347 | S-F | 327 | S-Cl | 271 |
S-Br | 218 | S-I | 170 | F-F | 159 | Cl-Cl | 243 |
Br-Br | 193 | I-I | 151 | C=C | 614 | C=N | 615 |
C=O | 745(799) | N=N | 418 | N=O | 607 | O=O | 498 |
C≡C | 839 | C≡N | 891 | N≡N | 945 |
3、键角(bond Angle)
键角即两共价键的夹角,由于共价键的方向性,共价化合物的键角是一定的,但组成相似的化合物未必有相同的键角,孤对电子对成键电子有较大的排斥作用,可导致键角变小。
4、键级(bond order)
键级是分子轨道提出的一个概念,其定义是成键电子与反键电子之差的一半,键级可以描述共价键的稳定性,键级越大,共价键越稳定。
5、键偶极矩(bond dipole moment)
键偶极距简称“键矩”,概念与力矩类似,可以描述共价键的极性。键矩的定义为:μ=q·l
式中μ为键矩(C·m),l为键长,q为电荷量
键矩是矢量,由电负性弱的一端指向电负性强的一端,即从正到负。键矩也可以由实验测得
分子模型
相比于键参数对共价键的描述,各种模型的描述显得更加直观。下表给出在分子模型中常用的颜色和对应元素。
元素 | 氧 | 碳 | 氮 | 硫 | 氢 | 氟 | 氯 | 溴 | |
|---|---|---|---|---|---|---|---|---|---|
用色 | 红 | 灰 | 蓝 | 黄 | 白 | 紫 | 黄绿 | 绿 | 橙 |
注:上表只是给出了常用的元素和对应颜色,与实际情况存在着一定的出入。
球棍模型(Ball-and-stick models)
与比例填充模型") 图11 甲烷的球棍模型(左)与比例填充模型
图11 甲烷的球棍模型(左)与比例填充模型
比例填充模型(Space-filling models)
比例填充模型与球棍模型类似,用来表现分子三维空间分布的分子模型。是球棒模型的进一步发展,可显示更为真实的分子外型。但很难从模型中看见化合物的键角。
存在范围
1.非金属单质
2.共价化合物
3.某些离子化合物