-
色谱图 编辑
色谱图是指被分离组分的检测信号随时间分布的图象。样品流经色谱柱和检测器,所得到的信号-时间曲线,又称色谱流出曲线。色谱图形状随色谱方法和检测记录的方式不同而不同,迎头色谱和顶替色谱的色谱图为一系列台阶;在洗脱法色谱中,若采用微分型检测器时,分离组分的检测信号随时间变化的图形为近似于高斯分布的一组色谱峰群,色谱图的纵坐标为检测器的响应信号,横坐标为时间、体积或距离。
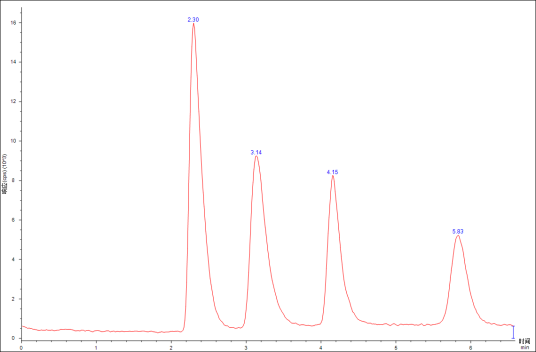
 图1 色谱图
图1 色谱图
基线
在实验操作条件下,当没有组分即仅流动相进入检测器时的流出曲线称为基线。
①稳定的基线:是一条水平直线,是测量基准,也是检查仪器工作是否正常的指标之一。
②基线噪声(baseline NOise):指由各种因素引起的基线起伏。
③基线漂移(baseline drift):指基线在一定时间内对原点产生的偏离,称为漂移。
色谱峰的高度、宽度
①峰高h:峰最高点至基线的垂直距离,称为峰高h,一般以mV或mm、cm表示。分析条件一定时,峰高是定量分析的依据。
②峰宽(peak width)或称区域宽度,它的大小反映色谱柱或所选色谱条件的好坏,峰宽有以下三种表示方法。
a. 标准偏差(stanDArd deviation)σ:标准偏差是指0.607倍峰高处色谱峰宽度的一半,图1中EF的一半,用σ表示。
b. 半峰宽(peak width at half-height)Y1/2:半峰宽是指峰高一半处色谱峰的宽度,图1中的GH,用Y1/2表示。
c. 峰底宽度(peak width at peak base)Y:峰底宽度是在流出曲线拐点处作切线,分别相交于基线上的I和J处之间的距离。常用Y表示。
标准偏差与峰底宽度和半峰宽的关系如下:
Y1/2=2σ
Y=4σ
色谱保留值
保留值表示试样中各组分在色谱柱中的滞留时间的数值,通常用时间或将组分带出色谱柱所需流动相的体积表示。
用时间表示的保留值反映被分离组分在色谱柱中的滞留时间,主要取决于它在两相间的分配过程,因而保留值是由色谱过程中的热力学因素所控制的。在一定的固定相和操作条件下,任何一种物质都有一个确定的保留值,该保留值可用作定性参数。
①死时间(dead time)tM :它是不被固定相吸附或溶解的组分(如气一液色谱的空气峰等)的保留时间(即不与固定相相互作用的组分,从进样开始到柱后出现浓度最大值时所需的时间,称为死时间),反映了流动相流过色谱系统所需的时间,因此也称为流动相保留时间。
②保留时间(retention time)tR:即组分从进样开始到柱后出现浓度最大值时所需的时间,称为该组分的保留时间。
③调整保留时间(adjusted rentention time)t'R:指扣除死时间后的保留时间。
t'R=tR-tM
保留时间是色谱法定性的基本依据。但同一情况下,同一组分的保留时间常受到流动相流速的影响,因此色谱工作者有时用保留体积来表示保留值。
④死体积(dead volume)VM:从进样器到检测器之间空隙体积的总和,包括色谱柱在填充后柱管内固定相颗粒间所剩留的空间、色谱仪中管路和连接头间的空间以及检测器的空间。当后两项很小且可以忽略不计,只考虑色谱柱中固定相颗粒间的空隙体积时,死体积可由死时间与色谱柱出口的载气体积流速F0来计算。
F0=tM×F0
式中,F0为在柱温为Tc(K)、柱出口压力为p0(MPa)时体积流量,mI·min-1。
体积流量是在柱出口处用皂膜流量计测定的。用这个方法测得的Fa值仅表示在室温下柱出口处的流量,这个值应该校正到柱温和干燥气体的情况下(排除皂膜流量计中水蒸气的影响):
F0=Fa×Tc/Ta×(p0-pw)/p0
式中,Tc为柱温,K;Ta为室温,K;p0为大气压力;pw为室温下水蒸气的分压。
⑤保留体积VR(retention volume):指从进样开始到柱后被测组分出现浓度极大值时所消耗流动相的体积。可由保留时间与色谱柱出口流动相体积流量的乘积来计算。
VR=tR×F0
当流动相流速F0加大时,保留时间tR相应降低,两者乘积仍为常数,因此VR与流动相速率无关。
⑥调整保留体积V'R(adjusted retention volume):只扣除死体积后的保留体积 。
色谱峰峰形异常问题
(1)色谱图中未出峰:进样问题(未进样或样品分解);流动相问题(泵未输液或流动相不正确),检测器设置不正确或检测器有问题。
(2)一个或几个峰是负峰:流动相吸收本底高;进入空气;离子对分离中的系统峰;样品组分的吸收(RI或UV)低于流动相。
(3)所有的峰均为负峰:信号电缆接反或检测器输出极性设置颠倒;光学装置尚未达到平衡。
(4)所有的峰都是宽峰:系统未达到平衡;溶样的溶剂比流动相强很多;色谱柱类型或尺寸不正确;色谱柱或保护柱被污染或降级;温度变化对色谱柱的影响。
(5)较早洗脱的峰呈宽峰:进样体积过大或样品浓度太高;进样器有问题,定量环(loop)大小不合适;在线过滤器、保护柱、色谱柱或管路堵塞;管路问题(内径不对或切管不正确);管路连接问题(接头或锥箍不正确);检测器时间常数不正确。
(6)峰比预想的要小:样品黏度过大;进样器有问题或进样体积有误;检测器设置不正确,定量环体积不正确;检测器输出未置零;用了不正确的检测器输出信号;检测池被污染;检测器的灯可能有问题。
(7)双峰或肩峰:进样量或样品浓度过大;保护柱或色谱柱进口堵塞;保护柱或色谱柱污染或失效。
(8)前伸峰:进样量或样品浓度过大;溶样的溶剂相对于流动相太强;保护柱或色谱柱污染或失效。
(9)拖尾峰:保护柱污染或色谱柱失效;进样问题;检测器时间常数不正确。
(10)平头峰:检测器设置不正确;进样体积太大或样品浓度太高。
(11)流动相洗涤强度南弱渐强时出现很多杂质峰:强溶剂将弱溶剂洗不出的杂质冲出来了,不影响柱的性能。
(12)在UV色谱图中,靠近死时间处出现负峰:进样时压力波动所致;样品溶剂比流动相的UV吸收值低。
进样量与峰面积不是线性关系
原因:样品在流动相中的溶解度小,只有部分样品被流动相冲入色谱柱中而另一部分则沉积在柱入口端。
实验的重复性差
原因:柱被污染,键合相流失,样品溶剂不同,样品稳定性不好,温度波动,流动相组成改变,缓冲液的pH值不合适,缓冲液的缓冲能力不足。
回收率低
原因:不可逆吸附.固定相过强或流动相过弱,非特异性吸附。
色谱峰保留时间问题
(1)保留时间飘忽不定(各次运行之间):系统不稳定或未达到平衡;泵压力不稳,有气泡;进样体积过大或浓度过大;温度波动;流动相混合不均匀;色谱柱被污染。
(2)保留时间增加或减少(各次运行之间):系统未达到平衡;泵流速变化;温度变化;柱污染,柱效下降;流动相被污染;溶剂入口过滤器堵或管路堵塞;系统渗漏。
(3)保留时间改变到一个新的恒定值:流动相不正确或其组成不正确;泵流速变化;实际输液的流速不正确(泵失灵或故障);温度变化;色谱柱尺寸或类型不正确;柱被污染;流动相含有稳定剂或稳定剂发生变化;输液系统的梯度滞后体积不正确 。