-
分子轨道理论 编辑
分子轨道理论又称分子轨道法(Molecular Orbital Theory)或MO法,1932年由美国化学家马利肯(R.S.Mulliken)及德国物理学家洪特(F.Hund)提出,是一种描述多原子分子中电子所处状态的方法。分子轨道理论是现代共价键理论之一,它的要点是:从分子的整体性来讨论分子的结构,认为原子形成分子后,电子不再属于个别的原子轨道,而是属于整个分子的分子轨道,分子轨道是多中心的;分子轨道由原子轨道组合而成,形成分子轨道时遵从能量近似原则、对称性一致(匹配)原则、最大重叠原则,即通常说的“成键三原则”;在分子中电子填充分子轨道的原则也服从能量最低原理、泡利不相容原理和洪特规则。
到了1933年,分子轨道理论已经被广泛的接受,并且被认为是一个有效而且有用的理论。事实上,根据德国物理化学家休克尔的描述,第一篇使用分子轨道理论的文献是由莱纳德琼斯发表于1929年。而第一个使用分子轨道理论的定量计算文献则是在1938年由库尔森发表的使用自洽场理论解决氢分子的电子波函数的工作。
到1950年,分子轨道彻底被定义为自洽场哈密顿算符的本征函数,这就是分子轨道理论发展成为一个严谨科学理论的标志。HF方法(Hartree-Fock method)是分子轨道理论的一种比较严谨的处理方法,尽管在一开始,HF方法是用来计算原子的电子结构的一种方法,但是在分子计算当中,分子轨道按照原子轨道的一组基集被拓展,发展出罗特汉方程,以此为基础,又发展出了各种各样的从头算量子化学计算方法。与此同时,分子轨道理论也被应用在了一种采用了更多近似方法的半经验计算当中,被称为半经验量子化学计算方法。
(1)在原子中,电子的运动只受1个原子核的作用,原子轨道是单核系统;而在分子中,电子则在所有原子核势场作用下运动,分子轨道是多核系统。
 分子轨道理论
分子轨道理论
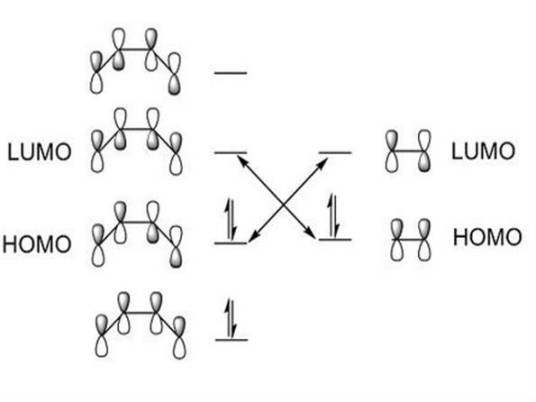
2、分子轨道可以由分子中原子轨道波函数的线性组合(linear combination of atomic orbitals,LCAO)而得到。有几个原子轨道就可以可组合成几个分子轨道,其中有一部分分子轨道分别由对称性匹配的两个原子轨道叠加而成,两核间电子的概率密度增大,其能量较原来的原子轨道能量低,有利于成键,称为成键分子轨道(bonding molecular orbital),如σ、π轨道(轴对称轨道);同时这些对称性匹配的两个原子轨道也会相减形成另一种分子轨道,结果是两核间电子的概率密度很小,其能量较原来的原子轨道能量高,不利于成键,称为反键分子轨道(antibonding molecular orbital),如 σ*、π* 轨道(镜面对称轨道,反键轨道的符号上常加“*”以与成键轨道区别)。还有一种特殊的情况是由于组成分子轨道的原子轨道的空间对称性不匹配,原子轨道没有有效重叠,组合得到的分子轨道的能量跟组合前的原子轨道能量没有明显差别,所得的分子轨道叫做非键分子轨道。
3、电子在分子轨道中的排布也遵守原子轨道电子排布的同样原则,即Pauli不相容原理、能量最低原理和Hund规则。具体排布时,应先知道分子轨道的能级顺序。当前这个顺序主要借助于分子光谱实验来确定。
对称性匹配原则
只有对称性匹配的原子轨道才能组合成分子轨道,这称为对称性匹配原则。原子轨道有s、p、d等各种类型,从它们的角度分布函数的几何图形可以看出,它们对于某些点、线、面等有着不同的空间对称性。对称性是否匹配,可根据两个原子轨道的角度分布图中波瓣的正、负号对于键轴(设为x轴)或对于含键轴的某一平面的对称性决定。
能量近似原则
在对称性匹配的原子轨道中,只有能量相近的原子轨道才能组合成有效的分子轨道,而且能量愈相近愈好,这称为能量近似原则。
轨道最大重叠原则
对称性匹配的两个原子轨道进行线性组合时,其重叠程度愈大,则组合成的分子轨道的能量愈低,所形成的化学键愈牢固,这称为轨道最大重叠原则。在上述三条原则中,对称性匹配原则是首要的,它决定原子轨道有无组合成分子轨道的可能性。能量近似原则和轨道最大重叠原则是在符合对称性匹配原则的前提下,决定分子轨道组合效率的问题。
上式是一种最粗糙的轨道近似,更好的近似是包含更多的原子轨道,这些原子轨道符合有效成键作用的三条件。例如,代替单纯的2s以及2pz的LCAO所形成的σ型分子轨道应为:
 ⑽
⑽
N2分子就属于这一类型。
有了式⑼与⑽的能级次序,就可按能量最低原理和泡利原理来预言同核双原子分子的基态(表3)。
表中的符号Σ、Π、… 意义与σ、π、… 相同,具有沿核间距方向角动量的含义,标志完整分子的态,由各个单电子轨道确定;右上角的+、-号指对平分两核的镜面反映为对称或反对称而言。
多原子分子的分子轨道 以上基于单电子波动方程近似解的轨道概念和方法,
 表3
表3
如前所述,分子轨道和能级是单电子波动方程的本征解,即满足:Hψi=εψi ⑿
式中H是单电子哈密顿算符,其中的位能描写一个电子在固定分子骨架及其余电子的平均作用。因而,H与其余电子的运动状态,即轨道有关。前面的讨论丝毫未触及H的具体形式,也未对分子轨道作过严格定义,所得结论是定性地适用的。为适应理论的定量化发展,已经推导出著名的哈特里-福克方程(见自洽场分子轨道法),对于闭壳层电子体系,式⑿中的H采取福克算符的形式:
|
 ⒀
⒀式中h是纯核场中单个电子的哈密顿算符,2Jj-Kj=Ji和2Jj(j≠i)代表其余电子的平均静电势,Kj(j≠i)称交换势能,它来源于泡利不相容原理导致自旋相同电子间的相关作用。Jj和Kj的表示式均明显地与分子轨道有关。
采用LCAO方法,分子轨道ψk 按式⑶表示成原子轨道φl(l=1,2,…,n)的线性组合:
 ⒁
⒁
代入⑿式,左右两端乘以φ奰并积分,求解归结为久期方程的本征值Ek和本征向量的自洽计算。哈特里-福克方程虽然较仔细地考虑了电子间的排斥作用,但由于平均势场模型仍然使一部分固有的“相关作用”未予考虑,因而理论计算结果仍未达到定量符合实验值的精度。改进的途径是考虑组态相互作用,已经出现了多种组态相互作用分子轨道从头计算程序,用于量子化学研究。
HF方法
Hartree-Fock SCF方法是一种从头算方法,而从头算方法简单的说,就是利用一个“正确的”哈密顿算符,除去最基本的常数之外,不再引用任何的实验数据,以薛定谔方程为基础,仅仅采用非相对论近似,Born-Oppenheimer尽速和单电子近似的基础上进行的薛定谔方程的求解和分子轨道的计算方法。
半经验计算方法
半经验法假定一个近似的哈密顿算符,并利用各种实验数据,如电离能、电子光谱的跃迁能、键能等数据,将积分的难度进一步简化,休克尔分子轨道理论(HMO)是这种方法的一个典型事例。
其他方法
还有诸如量子化学复合方法(Quantum chem-istry composite methods)、量子蒙特卡洛方法(Quantum MonteCarlo,QM)、组态相互作用(CI)、多组态自洽场方法(MCSCF)、多体微扰理论、耦合簇理(CC)等方法,这些是基于分子轨道理论发展起来的。
上一篇 环加成反应
下一篇 streptomyces