-
NF-kB信号通路 编辑
目录
NF-κB由Ranjan Sen(NIH)在诺贝尔奖获得者DAvid Baltimore的实验室中通过其与B细胞中免疫球蛋白轻链增强子中的II碱基对序列的相互作用而发现。
核因子-kB(NF-kB),是细胞内重要的核转录因子。它参与机体的炎症反应、免疫应答,能调节细胞凋亡、应激反应,NF-kB过度激活,与人类许多疾病如类风湿关节炎、心脏与脑部疾病的炎症变化等相关,因此通过药物来抑制NF-kB信号转导通路,可能会成为治疗的手段。
NF-kB分子的N端含Rel同源域,参与其和DNA结合、参与二聚体化,能被NF-kB抑制物(TeB)结合、抑制;NF-kB分子内还有核输出域、核定位域、转位活性域等,C端有反式转录激活域。P50/p65NF-KB能与靶基因启动子免疫球蛋白k轻链基因转录增强序列(kB序列)特异结合。RelA/c-Rel二聚体,能与靶基因启动子其他序列结合。
NF-kB家族有5个成员,包括NF-kB1(p50)、NF-kB2(p52)、RelA(p65)、RelB和c-Rel,通常所说的NF-kB蛋白,是指p65/p50亚单位形成的NF-KB1二聚体蛋白;RelB/p52亚单位形成NF-kB2二聚体蛋白。
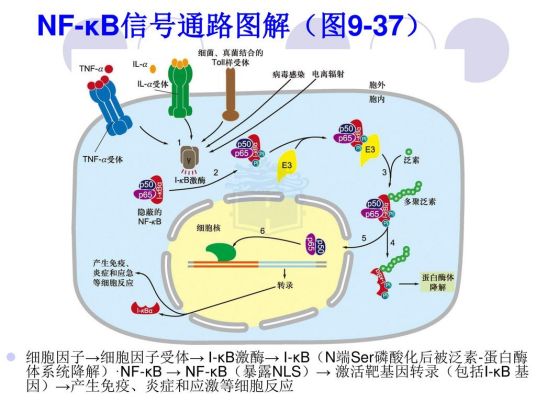
NF-kB可分两组:p50/p52组,分别由pII0、p105前体裂解产生,p50/p52能与NF-kB家族其他成员形成二聚体,存留于胞质。RelA(p65),RelB和cRel一组,没有前体。LkB是一种NF-kB的抑制蛋白,分子量36kD,可结合、抑制NF-kB并使NF-kB存留于胞质中,阻止NF-KB形成二聚体及入胞核,只能在胞质组成p50-p65-1eBa/p复合体。高水平肿瘤坏死因子a、佛波酯、脂多糖、白介素2、H2O2,等,可激活NF-kB诱导性丝裂原蛋白激酶,再将IkBa/β磷酸化,磷酸化的IkBa/β的Lys残基被泛素化后,可使IkBa/β降解,再使p50-p65NF-kB活化。
研究发现,激活NF-kB的信号转导通路主要有以下三种:经典通路、旁路通路和非典型通路。NF-kB1,RelA,c-Rel,均由经典通路激活,而NF-kB2,RelB,由旁路通路激活,此外还有DNA氧化损伤等诱导的非典型通路;p65的翻译后修饰,也调节NF-kB通路活性。
NF-kB信号通路激活的经典通路
当炎性因子肿瘤坏死因子a/白介素1/佛波酯/脂多糖等与相关受体结合后,引起后者构型改变,进而激活IkBa激酶,可使IkBa磷酸化,再在泛素连接酶P-TrCP的作用下泛素化,可被26S蛋白酶体识别并降解。于是NF-kB得以从细胞质NF-kB/IkBa复合物中释放出来,并活化、暴露核定位域,形成p50/RelA二聚体,迅速发生核转位,通过p50亚单位与靶基因的eB反应元件结合,从而启动靶基因表达如肿瘤坏死因子a和白介素1等,5分钟左右即可使细胞内NF-kB信号通路的活性水平达到峰值。蛋白激酶G/蛋白激酶C也可活化1kBa激酶、NF-kB
NF-kB信号通路激活的非经典通路
在肿瘤坏死因子受体TNFR家族配体,如CD40L,B细胞肿瘤坏死因子激活因子等的诱导下,相应的受体被激活,可促进NF-kB诱导的丝裂原蛋白激酶活化,再使蛋白激酶IKKa磷酸化,又使NF-KBp100被磷酸化降解,形成NF-eB的p52/RelB异源二聚体,又能使NF-eBp105被磷酸化降解,形成NF-kBp50/RelB异源二聚体,都进入细胞核从而调节靶基因的转录。CD40L,等还可活化NF-kB信号通路激活的经典通路。
NF-kB信号通路激活的其他通路
是指除了上述两种通路以外的其他通路的总称。适当水平的活性氧能活化酪氨酸激酶,再依次催化Rafl、蛋白激酶MAPKK/MAPK相继磷酸化,使蛋白激酶p90S6被激活,蛋白激酶p90S6有蛋白激酶IKK样活性,可使kBa的Tyr2残基磷酸化,结果1kBa被降解,NF-KB得以释放,继而发生核转位,与DNA上的kB反应元件结合,调节靶基因表达。该信号通路只在少数敏感性细胞中存在。
血管紧张素II可通过AT,R/活性氧通路及酪氨酸激酶Src/蛋白激酶P13K通路,激活NF-kB,上调白介素6、细胞间黏附分子-1等的表达,可促炎症、促进血管平滑肌细胞增殖。肿瘤坏死因子受体可通过肿瘤坏死因子受体相关因子(TRAF2/5/6)激活NF-KB,可促炎症,抗凋亡。Toll样受体信号通路活化、DNA损伤、UV辐射、蛋白激酶P13K、表皮生长因子受体(EGFR)、蛋白激酶C、内质网应激等,也可激活NF-wB通路。蛋白激酶A、蛋白激酶C,MSK1也可影响NF-cB活化的程度。p300/帽子结合蛋白CBP,p300/帽子结合蛋白CBP相关因子等,可使活化的NF-KBp50乙酰化,可增加NF-kBp50与DNA的结合力。NF-kB作用的靶基因有200多种,大致可分为以下六类,主要参与免疫应答、炎症反应、细胞增殖、抗凋亡、血管发生、肿瘤侵袭和转移等。rB反应元件存在于靶基因启动子中。
通过抑制IKK而抑制NF-KB的活性
将IkBa激酶-IKK的显性负突变体导入细胞中,可特异性阻断NF-cB的活化;针对NEMO(1KKy)的抗灸多肽,可与NEMO结合,从而阻止NEMO与IKKp的连接,可抑制急性炎症反应。小分子ATP竞争性抑制物,可抑制IKKp活性,进而抑制NF-kB活性,这为炎症反应的治疗提供了新方法。
通过kBa抑制NF-kB活性
这方面研究热点,是给予IkBa突变体、IcBa超级抑制剂,抑制NF-kB活性。
通过抑制蛋白酶体的活性抑制NF-KB活性
磷酸化的IkBa可被泛素蛋白酶体降解。泛素蛋白酶体抑制剂二肽硼酸类、广谱泛素蛋白酶体抑制剂MG-132,可抑制磷酸化的IkBa可被泛素蛋白酶体降解,能抑制NF-kB的活性。
通过对NF-kB的调控抑制NF-kB活性
对NF-KB的调控主要通过三个方面,即抑制其磷酸化、阻断其核定位及与DNA的结合、抑制靶基因表达。免疫抑制剂PG490,是从中药雷公藤中提取的一种二帖环氧化物,能选择性地作用于NFB的p65亚单位,抑制p65的转录激活域,在NF-KB与DNA结合之后,雷公藤可抑制靶基因的转录。一些药物如芍药苷,可抑制NF-kB的表达,从而可减轻由慢性脑血流灌注不足引起的脑损伤。合成与靶基因的顺式调控NF-KB元件相似的寡脱氧核糖核苷酸,将其转导入细胞,能在NF-kB入核前后,与NF-kB竞争,阻断NF-KB与靶基因启动子的结合活性,从而抑制靶基因的转录,可减轻炎症,已成功用于抑制炎症性休克,治疗再灌注损伤、心脑梗死等。
其他抑制NF-kB活性的策略
应用抗氧化剂,如N-乙酰多巴胺二聚体,可以抑制免疫和炎症反应中NF-kB的活性。探索具有靶细胞特异性和对不同NF-KB成员具有选择性的细胞内NF-kB活化阻断剂,可为临床治疗开辟新的途径。
研究发现,NF-kB调节的靶蛋白包括:
1.促凋亡因子,如Bax、胱冬蛋白酶II、CD95、死亡受体Fas、死亡配体FasL,GADD45g、癌蛋白-Myc,p5、肿瘤坏死因子受体1,TRAF结合蛋白TRAIL。
2.抑凋亡因子,如Bel-2,Bcl-xL,Bfl,c-FLIP、凋亡诱导蛋白IAPs,白介素13、肿瘤坏死因子a、肿瘤坏死因子受体1、肿瘤坏死因子受体相关因子TRAF1/2/6,IEX
3.细胞周期控制因子如癌蛋白cMyc,Rel、干扰索调控因子IRF-4,p21CIP1、周期索DI、周期素D2、周期素D3、ephrin-A1,GADD45β
4.生长因子,如白介素1/2/6/8/9/II/12/15、白介素2受体、正常T细胞活化下调蛋白RANTEs、粒细胞-巨噬细胞集落刺激因子。
5.黏附分子,如细胞间黏附分子(ICAM-1)/血管细胞间黏附分子(ICAM-V)/E-选择素。
6.促转移因子,如尿激酶型纤溶酶原激活物(u-PA)
7.促血管生长因子,如血管内皮生长因子(VEGF)
研究发现,NF-kB的诱导的受体包括:B细胞受体、T细胞受体、Toll样受体1~II、核苷酸结合域蛋白(NOD1)、肿瘤坏死因子受体1/2/4-1BB,Baff-R,CD27,CD30,CD40,死亡受体Fas/DR4/R5,外异蛋白A受体(EDAR),XEDAR,LTPR,OX40、白介素1受体、核因子受体活化蛋白受体RANK,RELT型肿瘤坏死因子受体、TIR、白介素18受体、Thelper1(THI)、补体C3a受体、补体C5a受体、KSHV-G蛋白耦联受体、表皮生长因子受体、整合素a5p1/a53/a6p4,p2整合素、趋化因子受体CXCR1/CXCR2/CXCR6、缓激肽受体(B2)、神经生长因子受体(p75,TrkA)、粒细胞一巨噬细胞集落刺激因子受体、血小板源性生长因子受体、溶血磷脂酸受体等。
癌症
NF-κB被真核细胞广泛用作控制细胞增殖和细胞存活的基因调节因子。因此,许多不同类型的人类肿瘤具有错误调节的NF-κB:即,NF-κB具有组成型活性。活性NF-κB启动基因的表达,使细胞保持增殖并保护细胞免受通过细胞凋亡导致其死亡的条件。在癌症中,控制NF-κB信号传导的蛋白质发生突变或异常表达,导致恶性细胞与其他生物体之间的协调缺陷。这在转移以及免疫系统对肿瘤的低效根除中都是明显的。当正常细胞从它们所属的组织中移除时,或当它们的基因组不能与组织功能协调地运作时,它们会死亡:这些事件依赖于NF-κB的反馈调节,并且在癌症中失败。
NF-κB的缺陷导致细胞凋亡的易感性增加,导致细胞死亡增加。这是因为NF-κB调节抗凋亡基因,特别是TRAF1和TRAF2,因此消除了caspase酶家族的活性,这是大多数细胞凋亡过程的核心。
在肿瘤细胞中,NF-κB具有活性(例如,41%的鼻咽癌),这是由于编码NF-κB转录因子本身的基因突变或控制NF-κB活性的基因(如IκB)基因);此外,一些肿瘤细胞分泌导致NF-κB活跃的因子。阻断NF-κB可导致肿瘤细胞停止增殖,死亡或对抗肿瘤剂的作用更敏感。因此,作为抗癌治疗的靶标,NF-κB是制药公司中许多活跃研究的主题。
然而,尽管令人信服的实验数据已经确定NF-κB是肿瘤发生的关键启动子,这为基于抑制NF-κB活性的抗肿瘤治疗的发展创造了坚实的理论基础,但在考虑抗NF时应谨慎行事。 -κB活性作为癌症治疗中的广泛治疗策略,因为数据还显示NF-κB活性增强肿瘤细胞对凋亡和衰老的敏感性。此外,已经显示经典NF-κB是Fas转录激活因子,而替代NF-κB是Fas转录抑制因子。因此,NF-κB促进癌细胞Fas介导的细胞凋亡,并因此抑制NF-κB的抑制可以Fas介导的细胞凋亡损害宿主免疫细胞介导的肿瘤抑制。
炎症
因为NF-κB控制着很多与炎症有关的基因,所以发现NF-κB在许多炎症性疾病中具有慢性活性也就不足为奇了,例如炎症性肠病,关节炎,败血症,胃炎,哮喘,动脉粥样硬化等。 。值得注意的是,一些NF-κB活化剂(如骨保护素(OPG))的升高与死亡率升高有关,尤其是心血管疾病。升高的NF-κB也与精神分裂症有关。最近,NF-κB活化被认为是香烟烟雾在骨骼肌中分解代谢作用的可能分子机制。肌肉减少症。研究表明炎症期间的细胞的功能取决于它的信号响应激活与相邻细胞和激素的组合,特别是通过特定的受体作用于细胞因子它接触。组织内的细胞表型通过反馈信号的相互刺激而发展,反馈信号与其他细胞协调其功能;当组织暴露于炎症时,这在细胞功能的重编程期间尤其明显,因为细胞改变它们的表型,并逐渐表达在消除炎症原因后准备组织再生的基因组合。特别重要的是在组织驻留细胞和免疫系统的循环细胞之间发展的反馈反应。不同细胞类型和免疫系统之间反馈反应的保真度取决于限制NF-κB激活的基因范围的机制的完整性,只允许表达有助于有效免疫反应的基因,并随后完成炎症消退后恢复组织功能。在癌症中,调节基因表达响应炎症刺激的机制被改变到细胞停止将其存活与协调其表型及其功能的机制与组织的其余部分联系起来的程度。这在NF-κB活性的严重受损调节中通常是明显的,其允许癌细胞表达NF-κB靶基因的异常群组。这不仅导致癌细胞异常运作:周围组织的细胞改变其功能并且仅停止支持生物体。另外,癌症微环境中的几种类型的细胞可以改变它们的表型以支持癌症生长。因此,炎症是一种测试组织成分保真度的过程,因为导致组织再生的过程需要协调不同细胞类型之间的基因表达。
NEMO
NEMO缺陷综合征是一种罕见的遗传病,与IKBKG中的缺陷有关,后者又激活NF-kB。它主要影响男性,并具有高度可变的症状和预后。
成瘾
NF-κB是ΔFosB的几种诱导的转录靶标之一,其促进发展和维持对刺激的成瘾。在尾壳核中,NF-κB诱导与运动增加有关,而在伏隔核中,NF-κB诱导通过奖励致敏增强药物的积极增强作用。